Neurodegeneration is a gradual loss of brain function that unfolds over decades before symptoms appear. It isn’t quite a silent killer like heart disease or cancer; it’s an emotional long goodbye. While neurodegeneration manifests in many ways, severe memory loss from dementia is among the most common outcome of this irreparable brain damage.
Each year, millions of individuals with dementia experience progressive losses in memory, personality, and functional independence. Countless family members and caregivers bear witness to this devastating life deterioration. Despite its toll on society, innovations combatting neurodegenerative disease have lagged far behind advances in heart disease that produced statins and the rapidly expanding arsenal of cancer-fighting drugs. These shortcomings are not due to lack of funding or interest from society, but instead because drug development for the brain faces complex challenges that other therapeutic areas do not.
Alzheimer’s Disease (AD) is the most common form of neurodegeneration and is responsible for ~70% of dementia cases. Over the past two years, the AD drug development space has started to turn a corner, highlighted by the first two disease modifying drug approvals. For the first time, patients have drugs that modestly slow cognitive decline, giving hope to both the patient and drug development communities.At Luma Group, we believe that these innovations are just the tip of the iceberg. The clinical learnings from AD on optimal diagnosis, delivery, and patient selection will catalyze further innovation in neurodegenerative proteinopathies like Parkinson’s disease, Lewy body dementia, Huntington’s disease, and amyotrophic lateral sclerosis (ALS). This whitepaper uses AD as a case study to explore the future of care in neurodegenerative disease. We present the current understanding of AD disease biology and treatment landscape for a generalist audience and give our vision of the next era of care in AD: a world where AD diagnosis is as easy as blood test and physicians can choose from a broad armamentarium of therapies to tackle the multifaceted features of neurodegeneration.
October 2025
What is Alzheimer’s Disease?
The defining hallmark of AD pathology in the brain is the presence of insoluble amyloid β plaques found outside of neurons and tau neurofibrillary tangles inside of neurons. The abnormal accumulation of these proteins has been the focus of AD drug discovery since Alois Alzheimer first reported on the disease in 1907. Alzheimer described a patient who “developed a rapid loss of memory” and was “disoriented in their own home.”1 This description matches our current understanding of AD clinical presentation, as patients become increasingly unaware of their surroundings, lose all ability to recognize family members, and eventually become prone to delusions and hallucinations.
At the molecular level, cognitive decline closely correlates with tau tangle pathology, but amyloid plaques appear in the earliest stages of AD. This suggests a sequential mechanism where amyloid initiate disease, but tau drives neurodegeneration. Beyond these two proteins, other factors, like chronic neuroinflammation in response to amyloid and tau, contribute to disease progression. Thus, while amyloid plaques and tau tangles are considered the hallmark pathology, AD is now recognized as a multifactorial disease in which amyloid, tau, and neuroinflammation interact to cause progressive neuronal damage and cognitive decline.2
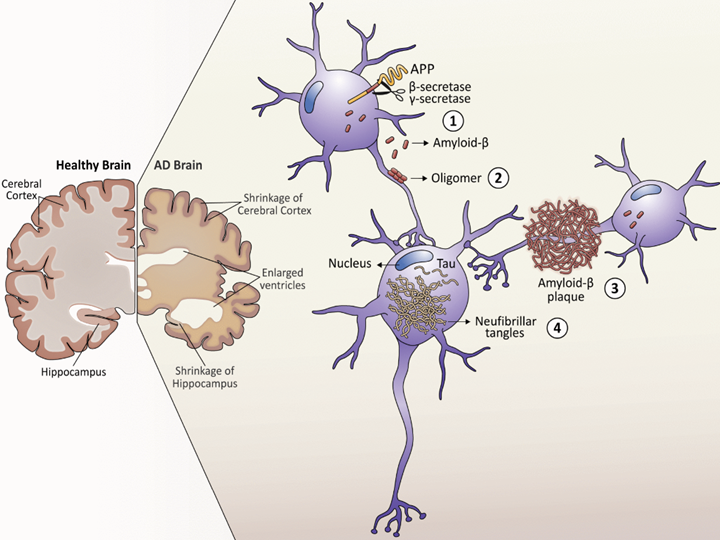
Figure 1: The sequential formation of amyloid plaques and tau tangles.
Source: Gomez W, Morales R, Maracaja-Coutinho V, et al. Down syndrome and Alzheimer’s disease: common molecular traits beyond the amyloid precursor protein. Aging (Albany NY) (2020 PMID: 31918411.
How are plaques and tangles formed?
Studies on amyloid processing have found that not all forms of amyloid β are toxic. Amyloid β is formed when two enzymes called β-secretase and γ-secretase cleave amyloid precursor protein (APP). Under normal conditions, these enzymes cleave APP to form soluble monomers of amyloid β, which occasionally make small aggregates that do not have any disease-causing properties. However, under AD conditions, these monomers aggregate into larger, insoluble amyloid fibrils and plaques that are a hallmark of AD, especially in early or presymptomatic AD. Tau neurofibrillary tangles on the other hand, tend to appear in the later stages of the disease. These are formed when native tau is modified with excessive phosphate groups (hypophosphorylation), which also makes tau more prone to aggregation.
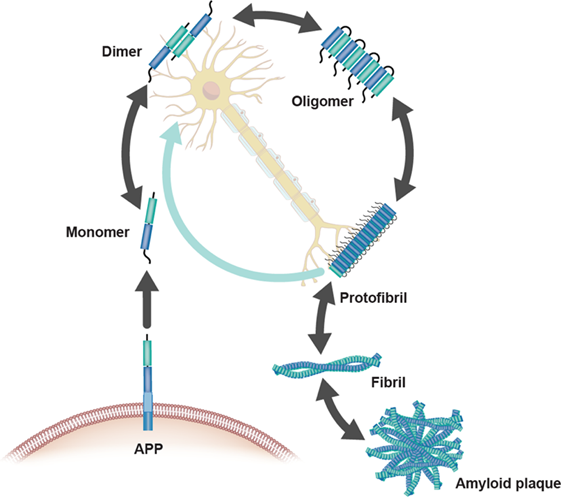
Figure 2: A closer look at the formation of insoluble amyloid plaques and fibrils
Source: Hampel, H, Hardy, J, Blennow, K et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol Psychiatry (2021). PMID: 34456336.
Why do plaques form?
Genetics research has provided some of the clearest clues on why these disease-initiating aggregates form. Mutations in the PSEN1 gene, a key component of the γ-secretase enzyme, alter the cleavage of APP and lead to production of different sizes of amyloid β monomers. Some of these monomers—particularly the amyloid β species that is 42 amino acids long—are more prone to aggregating into amyloid plaques.3 Importantly, PSEN1 mutations are the most common cause of early-onset AD, often leading to symptoms decades earlier than the typical age of onset. Nearly all patients with PSEN1 mutations have symptoms before the age of 60, with some case studies reporting symptoms as early as 32.4 While understanding PSEN1 disease biology offers important context on AD’s underlying mechanisms, autosomal dominant, inherited AD only accounts for ~1% of all AD cases. The majority of AD is classified as sporadic AD, caused by a mix of environmental and genetic risk factors that predispose you for disease. For example, aging is the strongest environmental risk factor for developing AD and certain forms of a gene called APOE can increase or decrease the chance of developing AD.5
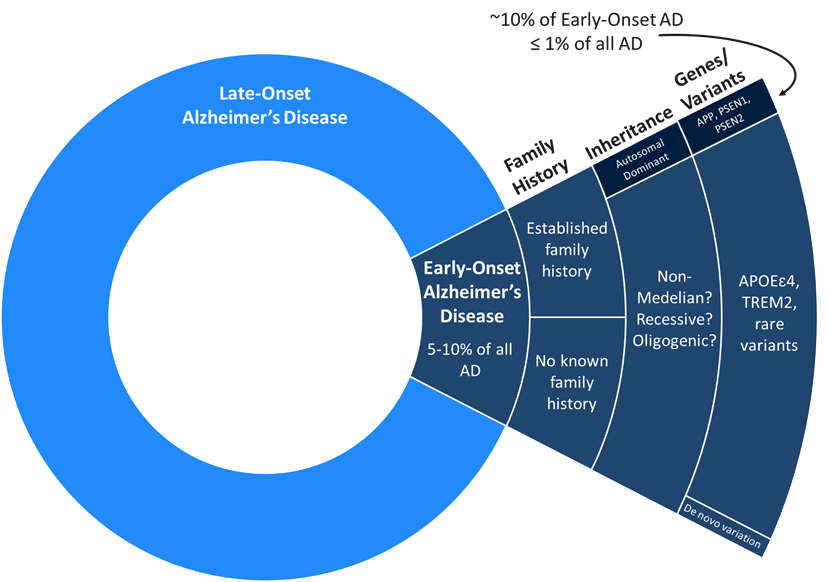
Figure 3: Prevalence of late-onset and early-onset AD
Source: Adapted from Sirkis DW, Bonham LW, Johnson TP, et al. Dissecting the clinical heterogeneity of early-onset Alzheimer’s disease. Mol Psychiatry (2022). PMID: 35393555.
Evidence of neuroinflammation in AD
In the age of deeper and cheaper DNA sequencing, new genetic discoveries have expanded our understanding of AD beyond the amyloid hypothesis. Notably, genome-wide association studies have implicated microglia, the brain’s resident immune cell, as a driver of AD symptoms. These studies found that mutations in a gene called “triggering receptor on myeloid cells 2” (TREM2) increases AD risk by 3-5x, fueling a new area of research to elucidate the role of microglia and neuroinflammation in AD disease progression.6, 7 The neuroinflammation hypothesis is relatively nascent, but our current understanding suggests that microglia are initially neuroprotective by eliminating plaques and tangles in the brain. However, as the production of amyloid and tau rapidly increases in later stages of disease, microglia fail to properly clear these proteins. Under this hypothesis, the microglia become chronically activated, creating a sustained pro-inflammatory environment in the brain that contributes to neuronal damage. Some emerging evidence from imaging studies in human patients has now indicated that the co-localization of amyloid, tau, and activated microglia in the brain are high risk-factors for cognitive impairment.8 Although the exact mechanisms linking neuroinflammation to AD pathology and neurodegeneration remain unclear, the involvement of microglia underscores that AD is a multifactorial disease, engaging many cell types and processes in the brain.
AD is now commonly grouped into a larger family of proteinopathies, where pathogenic proteins such as amyloid and tau are the hallmarks of the disease and drive symptom progression. As we discuss later, AD drug discovery efforts have primarily targeted amyloid β, either inhibiting its production or targeting it directly to stop the disease. Additionally, the emergence of the neuroinflammation hypothesis has also sparked new interest in microglia targeting therapies. Despite our growing understanding of AD biology, drug discovery has been long and challenging, and the first approvals of disease-modifying drugs that slow the progression of AD have only happened in the past two years despite decades of research efforts and failed trials.
Why is Alzheimer’s Disease drug discovery so challenging?
The central nervous system (CNS), a dense, intertwined network of neurons connected by innumerable synapses, is by far the most complex organ system in the human body. It is fundamental to our capacity to process information, experience emotion, and control movement. This intricate web is central to our survival, and the human brain is unique compared to other species, which complicates our ability to develop and test innovative therapies.
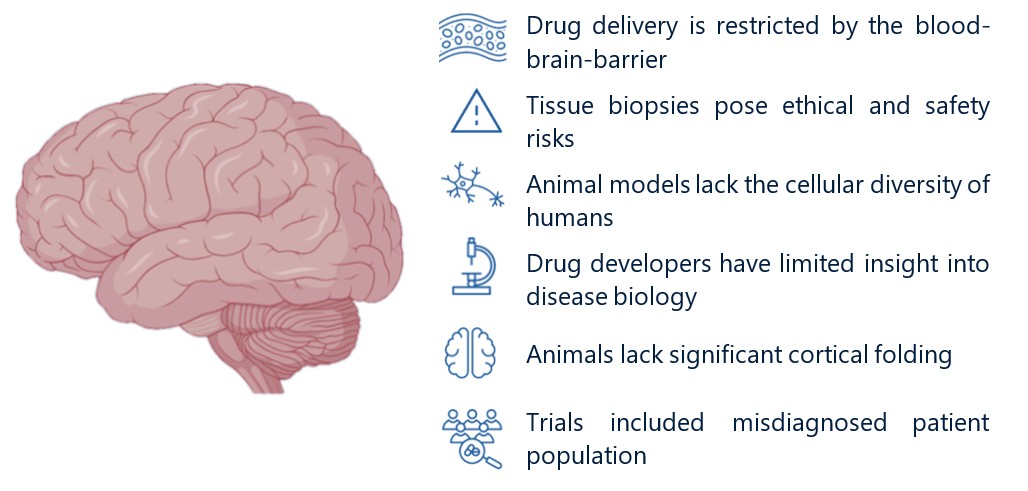
Figure 4: Challenges in CNS Drug Development
Source: Adapted from biorender.com
Biology differences limit predictivity of preclinical models
The human brain is uniquely complex compared to animal models (e.g., rodents, non-human primates, and others), resulting in low-fidelity preclinical studies that do not translate when we bring therapies into the clinic. While the human brain is notably larger compared to model species (even when you account for body size), this alone does not explain the lack of translatability. Equally as important, humans have developed a more structured cortex than other animals, with more cortical folding that increases our cortical density and surface area. At the cellular level, human brain cells are also more structurally complex and diverse: our neurons are longer and more interconnected (increased dendritic tree branching), and some human brain cells (e.g., interneurons, astrocytes, and glia) have much broader molecular diversity compared to animal models. Taken together, these differences help explain why current model systems fail to accurately recapitulate human brain function. Drug discovery is an iterative process that relies on rigorous preclinical studies conducted in animal models. However, because the brain lacks representative models, the path to developing AD therapies is especially difficult and high-risk.
Delivery challenges to the brain
CNS-targeted therapies must also overcome the restrictive blood-brain-barrier (BBB) to achieve relevant drug exposure levels. In organs like the liver, kidney, and heart, drugs transport freely from the blood into tissue. In the brain, however, drug penetrance is limited by the BBB, a tightly joined cellular layer that lines blood vessels in the brain and protects it from harmful pathogens. However, the BBB is also a formidable barrier for drug discovery, as over 98% of small molecules and nearly all antibodies fail to penetrate the BBB at therapeutically relevant levels. While all vertebrates have an intact BBB, many drugs tested in animal models fail to have comparable drug exposure levels when used in humans, adding a layer of additional complexity to CNS drug discovery. The BBB makes the CNS the most delivery-sensitive organ, and, combined with low-fidelity preclinical models, it is especially challenging to predict translatability of CNS therapies before entering clinical trials.
Clinical trial challenges
AD drug discovery has also been limited by the inability to only enroll patients with amyloid or tau pathology, confounding trial results. Until recently, AD patients were included in clinical trials based on questionnaires that assessed cognitive impairment. However, cognitive impairment is caused by many underlying conditions, and patients were often misdiagnosed with AD despite no evidence of amyloid or tau pathology. In early clinical trials, approximately 25% of patients were later found to have no amyloid pathology, even though these trials tested drugs that targeted the amyloid pathway.9
In recent years, the field has made many advancements using positron emission tomography (PET) imaging to positively confirm presence of amyloid and/or tau in living patients.10 Previously, amyloid and tau pathology diagnosis was only possible in post-mortem histological samples, limiting our ability to enrich clinical trials with patients who were positive for amyloid or tau. Using PET, patients are injected with a radiopharmaceutical tracer that makes amyloid or tau protein quantifiable using non-invasive imaging. With this well-validated method, we can now enroll patients who are positive for amyloid/tau pathology and can also stratify them based on the severity of their plaque burden. Given that modern AD drugs act directly on the amyloid pathway, PET has been a pivotal innovation that has pushed AD drug development forward.
The widespread adoption of PET imaging is reflective of the new age AD drug development, driven by novel diagnostics, new therapeutic innovations, and a deeper understanding of disease biology. As we will discuss in the next section, for the first time in history, patients have access to disease-modifying AD therapies. We are hopeful that this trend of novel diagnostic methods and transformative medicines will usher in a new era of neurodegenerative disease drug development, allowing us to precisely diagnose and treat the wide range of neurodegenerative proteinopathies, including AD, Parkinson’s Disease, dementia, ALS, and Huntington’s Disease.
History of Alzheimer’s Drug Development
The first AD therapies from the 1990s and early 2000s were approved for their ability to manage AD symptoms, not because they were disease-modifying. While they helped mask disease progression, the efficacy of these drugs wanes over time and they become less effective as neurodegeneration progresses. These early symptomatic therapies are key to delaying the onset of symptoms, but there was—and continues to be—great unmet need for a drug that slows or reverses disease progression.
| Category | Approved drugs | Target Neurotransmitter | Mechanism & Rationale |
| Cholinesterase Inhibitors | tacrine, donepezil, rivastigmine, galantamine | Acetylcholine | Increases acetylcholine signaling as patients show loss of cholinergic neurons11 |
| NMDA receptor antagonist | Memantine | Glutamate | Reduces neuron excitability by blocking glutamate signaling, as overactive excitability damages neurons11 |
Early drug development failures
The wealth of evidence implicating the amyloid β pathway has driven recent AD drug development efforts.12 The first attempts of disease-modifying drugs were inhibitors of γ-secretase and β-secretase (BACE1) activity, which act by decreasing total amyloid monomer production. However, these efforts largely failed. Semagacestat, for example, was the only γ-secretase inhibitor to progress to Phase 3 trials, where it failed to meet its primary endpoints, only modestly reduced amyloid levels, and cognitive decline slightly increased in the treatment group versus placebo.13 Similarly, the BACE1 inhibitors also failed to show improvements in cognitive decline relative to placebo, despite significant decreases in amyloid monomer production and moderate decreases in plaque burden.14 This is notable as many of the BACE1 inhibitor trials recruited patients with only very early or prodromal AD, suggesting that even the earliest symptomatic stages of AD may be too late to intervene with the secretase inhibitor class of drugs.
Turning a corner: The first clinical trial successes
The failure of the secretase inhibitors has narrowed the spotlight on monoclonal antibodies that directly bind to amyloid β. The first breakthrough came in 2021 with the accelerated approval of aducanumab (Aduhelm developed by Biogen). Lecanemab (Leqembi, developed jointly by Biogen, Eisai, and BioArctic) and donanemab (Kisunla developed by Eli Lilly) followed soon after. While aducanumab was eventually withdrawn from the market, lecanemab and donanemab were the first disease modifying AD drugs to receive full approval from the FDA. PET imaging played a key role in achieving these milestones, as it enabled rapid, non-invasive monitoring of disease modifying behavior, and was the biomarker that enabled use of the accelerated approval pathway. While these drugs have provided some validation of the amyloid hypothesis, they only show modest slowing of disease progression 15,16 and there is ample opportunity to build better and more efficacious AD therapies. Additionally, a new, amyloid-targeted treatment risk has emerged in the form of amyloid-related imaging abnormalities (ARIA) caused by vasogenic edema/effusions (ARIA-E) or microhemorrhages (ARIA-H). The exact cause of ARIA is still not clear, but these are dose-limiting side effects that exclude carriers of the ε4 allele of APOE from receiving lecanemab and donanemab, who are at increased risk of developing these side effects.
What does the future hold?
The progress of the AD drug development space in the last 25 years is undeniable, and the momentum to generate better disease modifying therapies will only continue. Moving forward, we foresee a world where AD and other neurodegenerative diseases are diagnosed using criteria beyond their symptomatic presentation. Instead, Parkinson’s Disease, ALS, Huntington’s Disease, and dementia will be defined as proteinopathies with distinct molecular signatures. These protein-level diagnoses will guide clinical recommendations of protein-specific treatment options, attacking the underlying pathology of neurodegenerative disease. With this framework in mind and driven by the current era of technological innovation in the life sciences, we envision multiple trends that are shaping the neurodegenerative treatment landscape for better patient outcomes:
- Diagnostics you can order from your primary care physician: Much like the implementation of PET tracers, the approval and broad adoption of blood-based diagnostics will be a meaningful step forward that benefits everyone a part of the AD patient journey. While PET is costly and requires recommendation of a specialist physician, blood-based diagnostics can be ordered by a primary care or community physician with a routine blood draw at a tenth of the cost. In the past year, both the Lumipulse (Fujibre) and Elecsys (Roche and Eli Lilly) blood-based diagnostics were approved by the FDA, with the Elecsys test being the first test approved for use in the primary care setting. Much like a routine lipid panel as part of a yearly checkup, we envision a future where patients can routinely access a “neurodegeneration panel” to screen the type of neurodegenerative disease or a multi-omics diagnostic to detect evidence of neuroinflammation or presymptomatic disease. Combined with new advances in AI/ML to deconvolute multifactorial signals, we look forward to seeing a future of early diagnosis of neurodegenerative disease and neuroinflammation, opening the door for prophylactic treatment of patients.
- Moving beyond amyloid and passive immunotherapy: While lecanemab and donanemab are passive immunotherapies (i.e., antibodies that bind to amyloid without an apparent mechanism that clears plaques), we are looking forward to innovations that actively eliminate pathogenic proteins and strategies that move to targets beyond amyloid, such as tau. The first targeted protein degrader (vepdegestrant developed by Arvinas and Pfizer) is scheduled to be approved in 1H 2026 for breast cancer. On the heels of this historic FDA decision, the time for novel mechanisms of action in neurodegenerative disease beyond passive amyloid clearing is on the horizon. While the current iteration of protein degraders have unfavorable drug-like properties that make brain penetrance challenging, we are closely following development of novel degrader approaches, including protein-based methods and modulators of autophagy, mitophagy, and other protein homeostasis pathways. As we move into this new era of viewing neurodegenerative disease as proteinopathies, we believe in innovative therapies that go beyond amyloid, using mechanisms of action that actively eliminate pathogenic proteins.
- Patient stratification that powers faster, smaller trials: Patients with neurodegenerative diseases face difficult outcomes with few disease modifying options. In the AD space, patients with PSEN and APP mutations (e.g., autosomal dominant AD) can see symptoms as early as their 30s. Estimates on the prevalence of autosomal dominant AD vary, but even conservative estimates suggest 10s of thousands of patients in the US.While these patients may be at highest risk of an aggressive disease course, they also have the clearest understanding of their underlying causes. The strong genetic nature of early-onset AD makes them especially addressable by gene therapies, and positive results from uniQure’s AMT-130 in Huntington’s Disease should serve as a beacon of innovation to follow suit in other neurodegenerative diseases.17 In a large patient population like AD, being able to enrich the treatment group with more homogenous biological underpinnings should attract innovation, not deter it, as these strategies open the door for faster, smaller trials that still have outsized impact in the patient community.
- Delivery that opens the gates of the BBB: Building off the foundational antibody approvals, there remains much work to improve their efficacy. The BBB typically restricts biologics to 0.1% or less of the injected dose into the brain and several efforts are leveraging receptor mediated transcytosis to shuttle biologics across the BBB, increasing brain penetrance by 10-100x. Roche’s trontinemab will be the first clinical validation of an active transport approach by targeting the transferrin receptor.18 Many other strategies have followed suit in this area, targeting receptors such as human CD98 and ALPL.19,20 Given that the CNS is the most delivery-sensitive therapeutic area, we are following how shuttle technologies can improve drug penetrance in the human brain, widening the therapeutic index of CNS-targeted therapies.
- Drug discovery using human brains in a dish: Due to the low predictive power of animal studies for CNS drug discovery, the field has turned to alternative models of human biology. Brain spheroid models are 3D in vitro models that utilize neuronal and non-neuronal cells from humans to create miniaturized human brains in a dish. While these models are far from perfect and can only recreate small networks of cells without an intact BBB, they offer an alternative, human-based model that can be combined with results from animal studies. Importantly, the FDA has published guidance that it plans to phase out animal studies in favor of alternative models like brain organoids and understanding how to best implement these models is a necessity for the field moving forward.21
- Resetting microglia to take back control of inflammation: With the neuroinflammation hypothesis taking hold in our understanding of AD, microglia targeted therapies are likely to play a major role in the AD treatment paradigm by synergizing with the amyloid therapies. Our current understanding of neurodegeneration has not definitively clarified the role of neuroinflammation in neuronal loss, but the preclinical evidence of an immune component in neurodegeneration grows daily. Moving forward, treatments that modulate microglia to control of neuroinflammation are likely to be necessary therapies in the armamentarium that can be used in combination to treat neurodegeneration.
- Next-generation therapies progressing into the clinic: The therapeutic landscape for CNS diseases is nascent and new technological innovations are continuing to make their way into the brains of patients. In this next era of care for neurodegenerative diseases, we will see these therapies enter the clinic at scale, pushing the boundaries of how we think about patient treatment options. In Parkinson’s disease, we have already seen promising clinical data using pluripotent stem cell therapy.22 We are excited to see the continuation of these technological discoveries, including alternative cell therapies and strategies for cellular reprogramming, enter the patient treatment paradigm.
The next era of caring for neurodegenerative disease
Given these areas of core innovation that we are likely to see in the near term, we envision a multi-pronged approach in the future that gives patients with AD and other neurodegenerative diseases a multitude of options to address disease head on. Diagnostics will be the foundation of this new age of medicine, giving us predictive insight to identify the disease before symptom onset. In a perfect world, patients as young as their 40s will be empowered to take a panel of blood-based and genetic diagnostics, without the fear that their diagnosis will mean a slow-moving death sentence. Abolishing that fear will require disease modifying drugs that actively address the pathogenic underpinnings of disease, that are safe to use chronically and prophylactically. While this is a far cry from where we stand today, we believe that the innovations on the horizon will be the first steps to make this vision a reality.
- Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int ↩︎
- Heneka, MT, van der Flier, WM, Jessen, F et al. Neuroinflammation in Alzheimer disease. Nat Rev Immunol (2025). PMID: 39653749. ↩︎
- Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int J Mol Sci (2023). PMID: 36835161. ↩︎
- Andrade-Guerrero J, Santiago-Balmaseda A, Jeronimo-Aguilar P, et al. Alzheimer’s Disease: An Updated Overview of Its Genetics. Int J Mol Sci (2023). PMID: 36835161. ↩︎
- Eid A, Mhatre I, Richardson JR. Gene-environment interactions in Alzheimer’s disease: A potential path to precision medicine. Pharmacol Ther (2019. PMID: 30877021. ↩︎
- Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med (2013). PMID: 23150908. ↩︎
- Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med (2013). PMID: 23150934; PMCID: PMC3631573. ↩︎
- Pascoal TA, Benedet AL, Ashton NJ, et al. Microglial activation and tau propagate jointly across Braak stages. Nat Med (2021). PMID: 34446931. ↩︎
- Karran E, Hardy J. Antiamyloid therapy for Alzheimer’s disease–are we on the right road? N Engl J Med (2014). PMID: 24450897. ↩︎
- Chapleau M, Iaccarino L, Soleimani-Meigooni D, et al. The Role of Amyloid PET in Imaging Neurodegenerative Disorders: A Review. J Nucl Med (2022). PMID: 35649652. ↩︎
- Raina P, Santaguida P, Ismaila A, et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann Intern Med (2008). PMID: 18316756. ↩︎
- Karran E, De Strooper B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov (2022). PMID: 35177833. ↩︎
- Doody RS, Raman R, Farlow M, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med (2013). PMID: 23883379. ↩︎
- Egan MF, Kost J, Voss T, et al. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N Engl J Med (2019. PMID: 30970186. ↩︎
- Mintun MA, Lo AC, Duggan Evans C, et al. Donanemab in Early Alzheimer’s Disease. N Engl J Med (2021). PMID: 33720637. ↩︎
- van Dyck CH, Swanson CJ, Aisen P, et al. Lecanemab in Early Alzheimer’s Disease. N Engl J Med (2023). PMID: 36449413. ↩︎
- https://www.uniqure.com/investors-media/press-releases ↩︎
- Grimm HP, Schumacher V, Schäfer M, et al. Delivery of the Brainshuttle™ amyloid-beta antibody fusion trontinemab to non-human primate brain and projected efficacious dose regimens in humans. Mabs (2023). PMID: 37823690. ↩︎
- Chew KS, Wells RC, Moshkforoush A, et al. CD98hc is a target for brain delivery of biotherapeutics. Nat Commun (2023). PMID: 37598178. ↩︎
- Voyager Therapeutics, Corporate Presentation (October 2025) https://ir.voyagertherapeutics.com/static-files/329b71f5-f944-496c-b757-372a92e82b55 ↩︎
- https://www.fda.gov/news-events/press-announcements/fda-announces-plan-phase-out-animal-testing-requirement-monoclonal-antibodies-and-other-drugs ↩︎
- Tabar V, Sarva H, Lozano AM, et al. Phase I trial of hES cell-derived dopaminergic neurons for Parkinson’s disease. Nature (2025). PMID: 40240592. ↩︎